Sleeplessness - Here is how
Nov 10, 2024

This is Nikolai Tesla. One of humanity's sharpest minds, he functions on just 4 hours of sleep. It sounds like sc-fi but thanks to agene mutation (DEC2), a few lucky people can already do this. They are more wakeful, more energetic, and even have enhanced attention, motivation and even leaner body mass (Source, Source, Source). The best part: they do not suffer from daytime sleepiness or the consequences of sleep deprivation - instead they wake up after 4-6 hours feeling well-rested.
We're two grad students @Harvard and @MIT working to make this a reality for everyone. Here is our roadmap:
First, How does it work?
It’s simple: Individuals with familial short sleep have different mutations that disrupt the DEC2 gene (Source). This gene mutation increases levels of orexin—a key wakefulness-promoting neuropeptide—which leads to reduced sleep need and increased energy without feeling sleep-deprived.
Firstly, we already know it works in humans (Source, Source). This cannot be highlighted enough: We are not making anything up. This already works. Secondly, a group has shown that it works in young mice by simply inserting the mutation via plasmids (Source). But until today, you can only either inherit it, if you are lucky enough to win the genetic lottery, or if you are young enough to possibly use the previous approach in mice. But that won’t solve the problem of the millions of adults struggling with low energy, fatigue, and other sleep issues.
We need something to mimic this effect at any age.
Idea 1: Using Prime Editors to Replicate the Safe and Proven Phenotype Observed in Humans
As of now, prime editing is a non-toxic, highly effective, precise way (Source, Source, Source) to introduce the genotype seen in humans. A specific mutation identified in humans is suitable for Cytosine Base editors (c.1086C>T, p.Tyr362His) (Source).
Yet, a couple of challenges exist:
Problem 1) Effective Delivery
How to get the prime editors to target only the cells that have the DEC2 gene (check if orexin-DEC2 connection has been explained)? State-of-the-art AAV vectors offer robust, non-invasive gene delivery with low immunogenicity and have been shown to work for gene alterations in the brain, overcoming the hurdle of crossing the blood-brain barrier (Source). The challenge is that they have a limited capacity load of 4.7kB and won’t fit a prime editor. David Liu’s lab, which famously invented prime editors, has demonstrated a potential solution: Dual AAV vectors where Vector A fits the genetic sequence and Vector B the editor demonstrates up to 59% editing efficiency in the mouse brain (Source) and others have additionally shown proof of concept (Source, Source, Source)
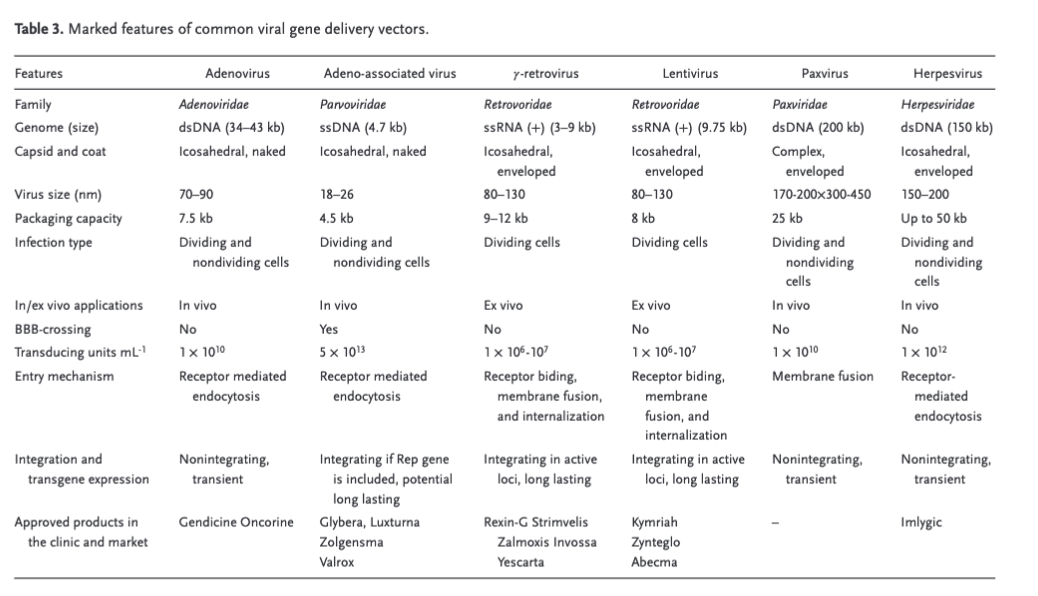
Source Engineering Gene Therapy: Advances and Barriers
Problem 2: Specificity
If anything is inserted into the brain, it should only target the specific site in the brain, namely the orexin-producing cells in the hyptothalamic suprachiasmatic nucleus (SCN). The more specific, the fewer side effects.
Solution: We need a sensor that scans the cells like barcodes, identifying the neurons we want to target. Luckily, something like this exists: RNA sensors (Source) that can detect whether a cell is translating RNA unique to that cell type. As our target cells in the hypothalamus perform specific task, they have distinct RNAs that can be used as markers, such as these from Vasoactive Intestinal Peptide [VIP] (Source) and Arginine Vasopressin (AVP) (Source). If this RNA is detected, then initiate prime editing; otherwise, don’t.
Challenge 3: Blood-Brain Barrier
Ideally, we want this to be an non-invasive application that crosses the blood-brain barrier. Luckily, tons of research exists in this field (Source). The hypothalamus, our target organ, is a circumventricular organ with highly permeable capillaries—that’s good. AAVs are roughly 20nm in size (Source), so they can potentially pass through these capillary fenestrations, which range from 60-70nm (Source).
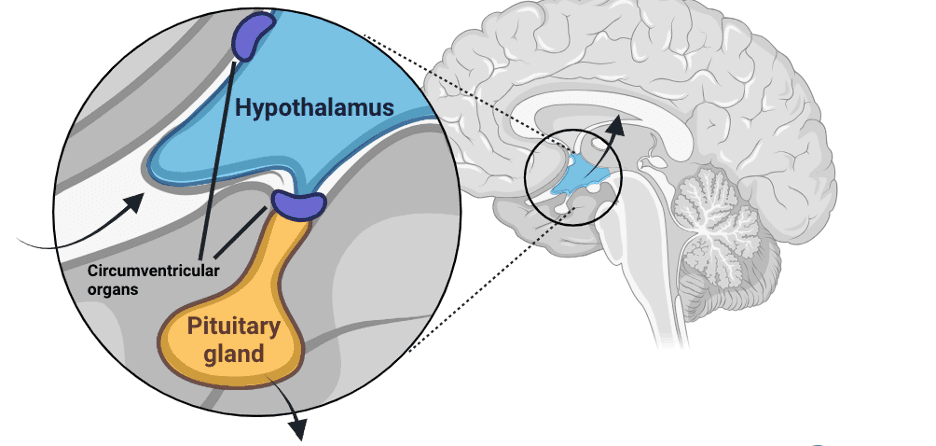
Second, AAV viruses can be engineered to ease this project, e.g., engineering AAVs to facilitate transcytosis or engineering capsids with receptors that enable crossing the blood-brain barrier (Source, Source). AAV9 has been shown to deliver gene therapies to the brain and has demonstrated safety in studies (Source). A completely different approach would involve opening the blood-brain barrier using focused ultrasound (Source). This method works by inducing microbubble oscillation within cerebral blood vessels, disrupting tight junctions between endothelial cells, and allowing therapeutic agents to pass through the BBB into brain tissue. Recently, promising data for intrasnal application of AAV viruses (Source) has been shown, allowing retrograde delivery through the olfactory bulb - tons of things to try.
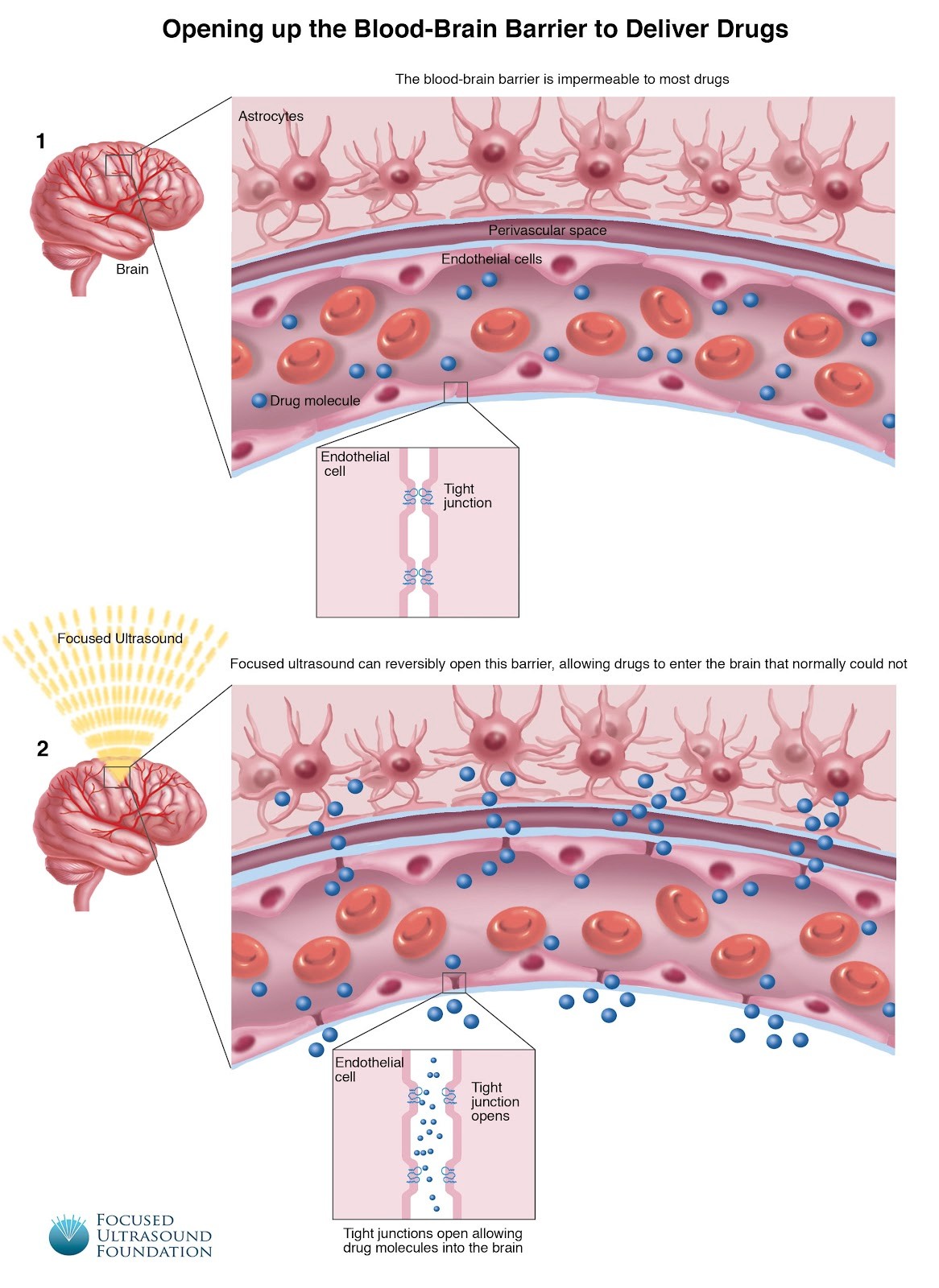
Image: Blood-Brain Barrier Opening by Focused Ultrasound Foundation
Lastly, we might not even need a highly penetrative virus. Since the protein’s mRNA expression is only slightly lowered in the original mutation, small effect sizes would be sufficent (Source).
Idea 2: Modulate Gene Expression at the RNA Level
A less invasive version would consist of Antisense oligonucleotides (ASOs) targeting DEC2 mRNA and preventing it’s translation. This offers precise mRNA degradation with potentially low side effects (Source). Unlike gene editing, ASOs offer reversible effects, making them a safer choice (Source). Their smaller size (between 15-25 nuelcotides) make it more feasible to be inserted into the brain (Source).
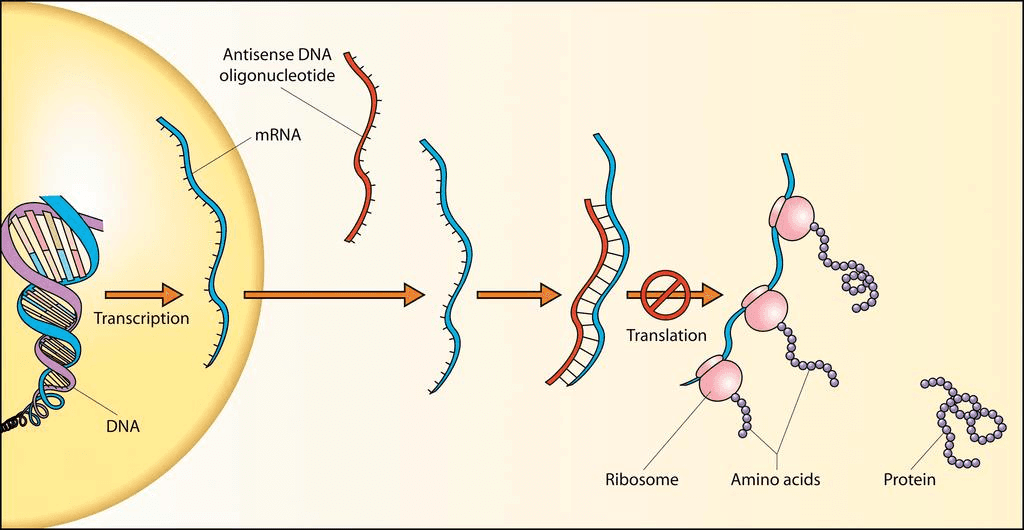
Additionally, ASOs have been proven effective and safe in clinical settings; for example, the therapy Spinraza (Source), used for spinal muscular atrophy has shown promising results with minimal side effects.
Idea 3: DEC 2 Protein Degradation
A different alternative would involve simply degrading the DEC2 protein by activating the cell’s own autophagy system through Targeted Protein Degradation (TPD) (Source).
How this works:
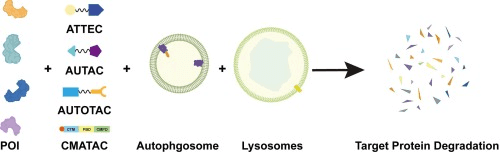
Source: Autophagy mediated targeting degradation, a promising strategy in drug development
To ensure that the autophagy system recognizes and degrades the correct protein, DEC2 would need a tag signal for degradation. Various methods include Autophagy-Targeting Chimeras (AUTOTACs) (Source), which link a recognizable structure on our target molecule to an autophagy-inducing ligand (Source). Steps include:
1. Design a ligand that binds to the target protein.
2. Choose an autophagy-targeting motif, e.g., a sequence that interacts with LC3, to initiate autophagy.
3. Link these elements together to guide DEC2 to the autophagic pathway.
There are multiple approaches for targeting protein degradation, other ideas include:
MethodHow It WorksAUTACsDirects proteins to autophagy using bifunctional moleculeLYTACsTags extracellular proteins for lysosomal degradationMolecular GluesPromotes interaction with autophagy receptorsCRISPR/Cas9 TaggingUses small molecules to add autophagy tagsChemical ModificationUses KFERQ-like motifs recognized by chaperones
Idea 4: Direct Inhibition of DEC2 Function via small Molecules
If DEC2 has an active site or binding pocket, one could also try directly inhibiting DEC2 via small compounds. The main challenge would be to find a suitable molecule. For that, one can use high-througput screening to test small molecule libraries to identify inhibitors that bind to and disrupt its activity. There has been tons of progress in the field, such as ultra large virtual screens (Source) and recently even AI enabled virtual screening of vast chemical libraries (Source)
Idea 3 and 4 offer low toxicity but raises questions about the full degradation of DEC2 compared to the specific mutations observed in humans. Full DEC2 degradation could have unknown downstream effects on orexin pathways or, conversely, enhance the positive effects by reducing sleep need further—this is worth exploring.
Tons of other ideas exist. Here are a couple more:
Detailed View:
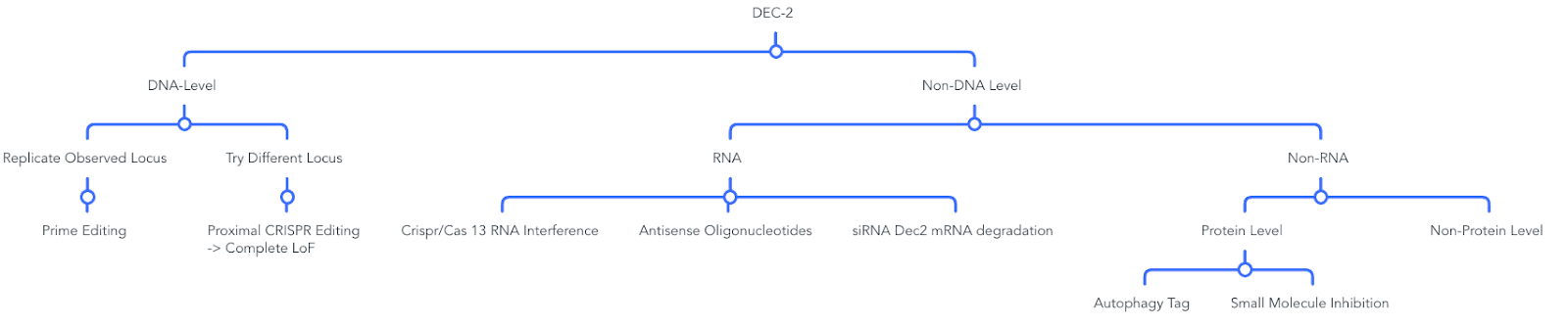
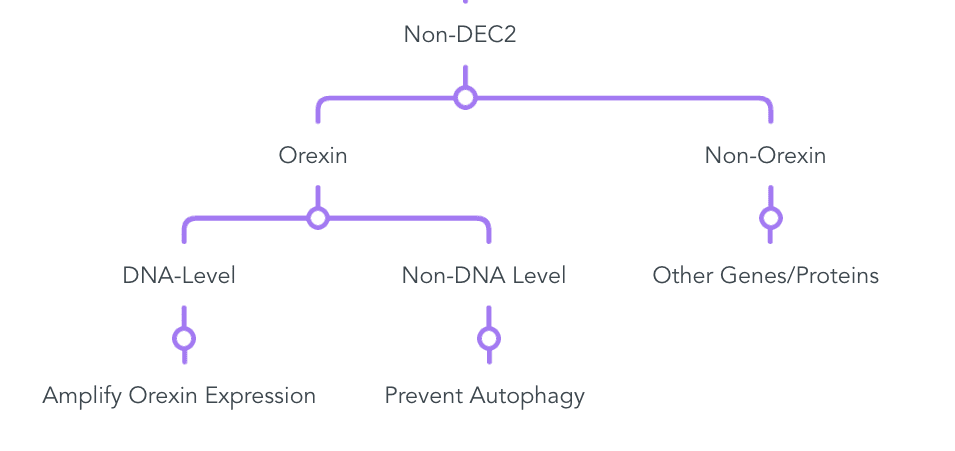
Most important to remember: This phenotype already exists. It works and is safe. The only question is how to mimic this in the easiest way. It’s not about if, just how.